

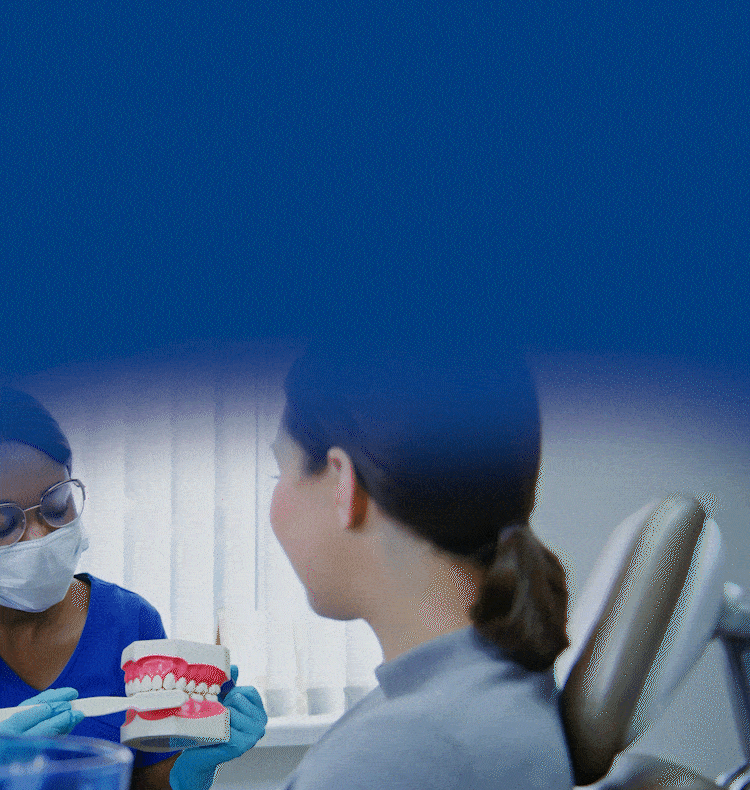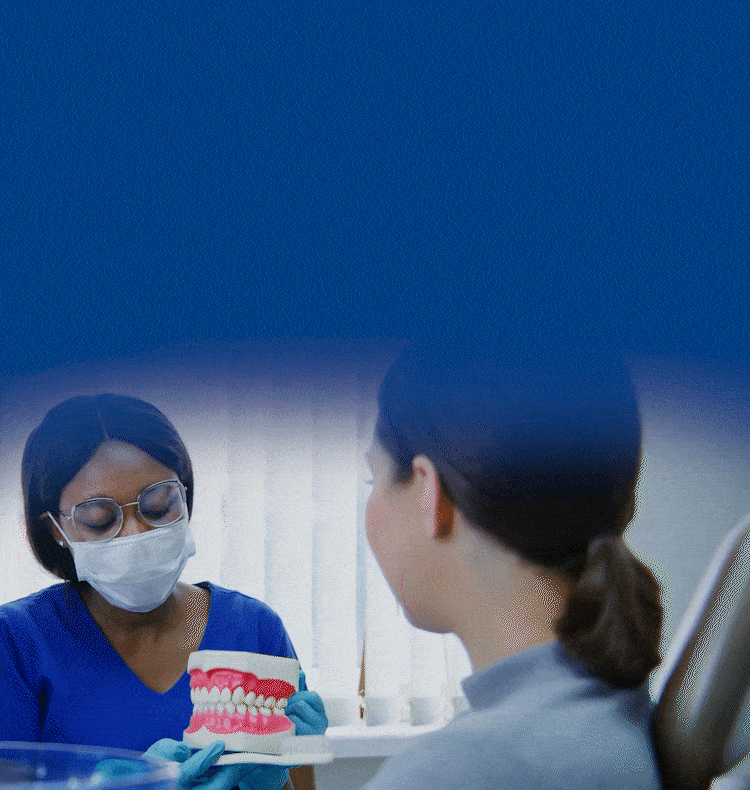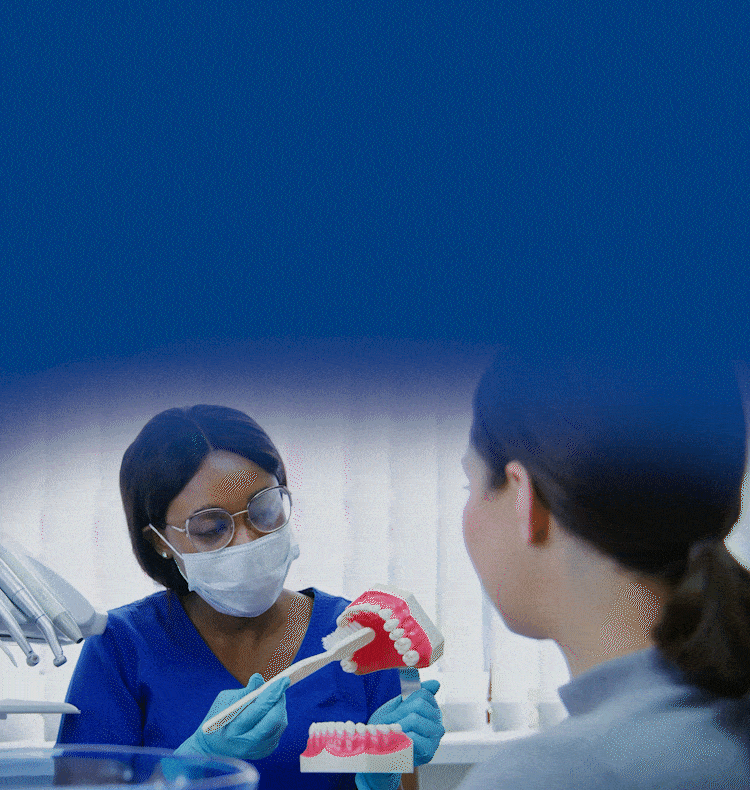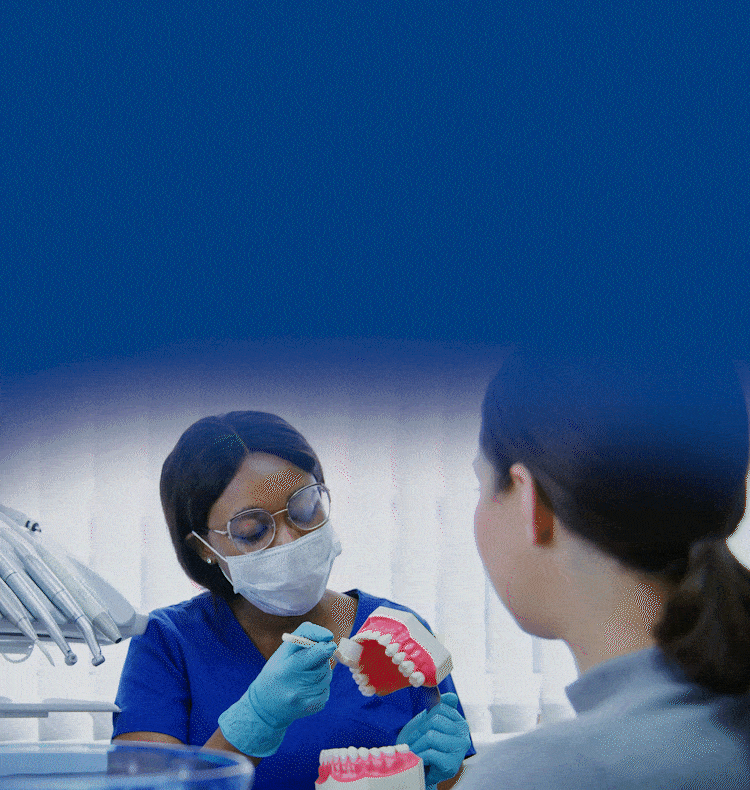
TOGETHER WE CAN BUILD HEALTHIER & BRIGHTER SMILES
150+ FREE Continuing Education Courses
Featured Courses


Explore our products on Crest + Oral-B ProShop
Take advantage of professional exclusive pricing for your office and patients’ needs or your personal professional use.












Crest Pro-Health Gum Detoxify
Order Now
Crest Kids Color Changing Toothpaste
Order Now
Crest Pro-Health Gum Detoxify Toothpaste Sample Size
Order Now
Clinically Proven Healthier Gums
Learn More







Professional Resources

Oral-B iO - Why recommend oscillating rotating technology?
Learn More
Whole Body Health
Learn More
#Hygienist Proud - Resources for you!
Learn MoreStudent & Faculty Quick Links
Enter Assignment Number
Use the Assignment Number provided to find the course
Create assignments with a wide range of courses available
Review the submitted Assignments
Case studies for students and professionals
Interactive learnings for you